Qiu Lab Meetings
Jump to navigation
Jump to search
Projects & Goals
- Borrelia population genomics: Recombination & Natural Selection (Published)
- Borrelia pan-genomics (Submitted)
- Positive and negative selection in Borrelia ORFs and IGS (In submission)
- Dr Bargonetti's project (Summer 2013)
- A population genomics pipeline using MUGSY-FastTree (Summer 2013): Project page
- Borrelia Genome Database & Browser (Summer 2013) Version 2 screen shot
- Pseudomonas population genomics (Summer 2013) Project page
- Hypothesis Testing: Do host-interacting genes show adaptive codon usage? (Summer 2013): Project page
- Phylogenomics browsing with JavaScript/JQuery, Ajax, and jsPhylosvg
- Frequency distribution of ospC types in wild tick populations (Fall 2013) Project page
{kind=link}
Lab meeting: June 13, 2013
- Weigang: IGS paper submission should be done by Thursday.
- Che/Slav: Workshop update (Meeting at 3:30pm?)
- Che: SILAC project (Meeting at 4pm?)
- Zhenmao: Tick processing & paired-end Illumina sequencing
- Pedro: Updates on "ncbi-orf" table
- Girish: phyloSVG extension; QuBi video
- Saymon and Deidre: consensus start-codons
- Reeyes and Raymond: Pseudomonas DB; fleN alignment and phylogeny
- Valentyna: BLASTn results (4:30pm?)
Lab meeting: May 23, 2013
- May 24, Friday: End of School Year Party in the Park (we leave from Hunter @ 1:30pm)
- Recommended reading of the week: Detecting Neanderthal genes using the D' homoplasy statistic
- Weigang: IGS paper submission
- Che: Thesis update/SILAC project/Summer teaching
- Zhenmao: Manuscript update: Material & Methods; Results (Tables and Figures)
- Pedro: Catlyst web framework
- Girish: cp26 phylogenomic analysis
- Saymon and Deidre: consensus start-codons
Lab meeting: May 16, 2013
- Weigang: IGS paper submitted yet?
- Che: Thesis update. Chapter 3. Evolution of ospA/ospB gene family
- Pedro/Zhenmao: Can we wrap up the BLAST identification of ospC types?
- Girish: Fetch cp26 sequences from DB; Run MUGSY & FastTree
- Saymon/Deidre: Identification of consensus start-codon positions
- Pedro/Girish: orth_get/orth_igs website development. Catalyst. Implement graphics (genome map & phylogeny) query interface
- Raymond: start the Pseudomonas summer project
Foundational Readings
- Molecular phylogenetics
- Population genetics
- Genomics
- Systems Biology
Informatics Architecture
- Operating Systems: Linux OS/Ubuntu, Mac OS
- Programming languages: BASH, Perl/BioPerl, R
- Relational Databases: PostgreSQL
- Software architecture
- bb2: Borrelia Genome Database
- bb2i: an Perl API for bb2
- DNATweezer: Perl wrappers of most frequently used BioPerl modules, including Bio::Seq, Bio::SimpleAlign, and Bio::Tree [1]
- SimBac: A Perl/Moose package for simulating bacterial genome evolution [2]
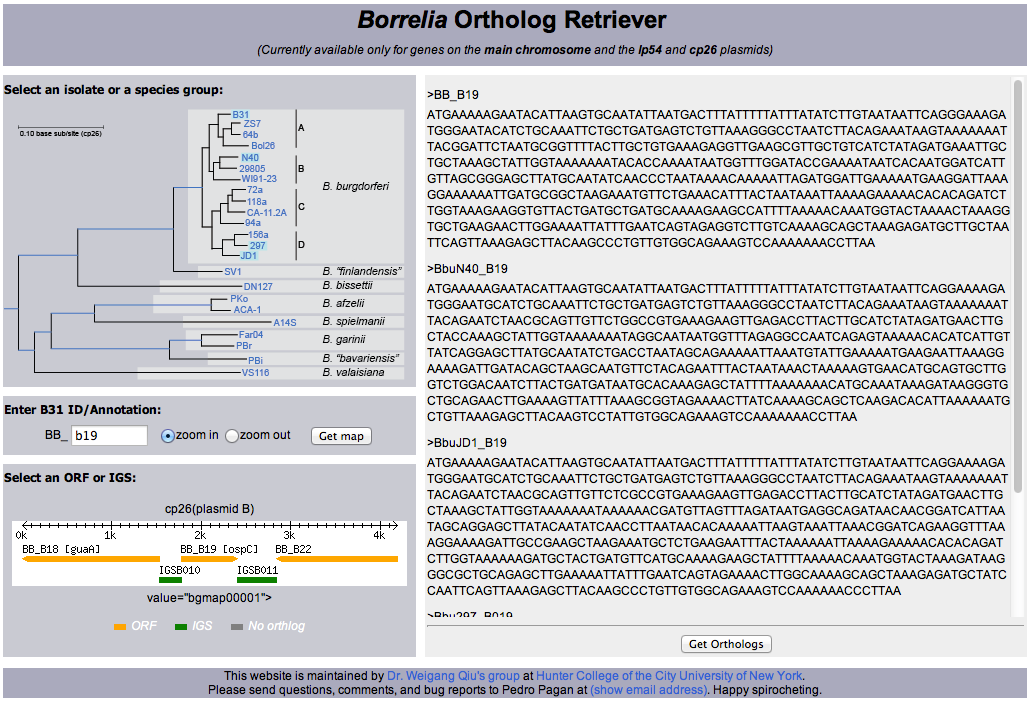
- Borrelia Ortholog Retriever: Download ortholog alignments from 23 Borrelia spp genomes. Search by gene names and IDs.[3]
- Hardware Setup
- NSF File Server
- Database and Application Server
- Web Server
- Linux Workstations
Perl Challenges
| Problem | Input | Output |
|---|---|---|
| DNA transcription | A DNA sequence, in 5'-3' direction (e.g., aaatttaaaagacaaaaagactgctctaagtcttgaaaatttggttttcaaagatgat) | An RNA sequence, in 5'-3' direction |
| Genetic code | None | 64 codons, one per line (using loops) |
| Random sequence 1 | None | Generate a random DNA sequence (e.g., 1000 bases) with equal base frequencies |
| Random sequence 2 | None | Generate a random DNA sequence with biased base frequencies, e.g., 10% G, 10% C, 40% T, and 40% A. |
| Graphics I | a categorical dataset, e.g., Biology | a bar graph & a pie char, using GD::Simple or Postscript::Simple |